MatSQ has modularized each function, making it easy to add and modify functions.
 Structure Builder is a powerful, intuitive module that allows you to create, visualize, and manipulate DFT/MD simulation models such as crystal structures, molecules, and monomers.
Structure Builder is a powerful, intuitive module that allows you to create, visualize, and manipulate DFT/MD simulation models such as crystal structures, molecules, and monomers.
Structure Builder consists of the visualizer menu that displays the structure and the manipulator menu for manipulating structures.
The simplest way to create a simulation model is to upload a structure file.
Drag and drop a file to Visualizer or click on the icon to upload a file. You can upload most file types, such as CIF, POSCAR, PDB, xyz, and so on. Alternatively, you can add structures using the 'Modelling' tab menu.
icon to upload a file. You can upload most file types, such as CIF, POSCAR, PDB, xyz, and so on. Alternatively, you can add structures using the 'Modelling' tab menu.
You can use several functions by manipulating the mouse on the structure canvas. Moreover, you can use the mouse wheel to zoom in/out or drag to rotate the time point. Right-click to use the following functions:
MatSQ provides a variety of tools to manipulate the atomic structure. You can also use the Manipulator menu to add or move atoms, resize cells, or clone unit cells to create supercells. Up to 10 Manipulate details are stored in the history at the bottom of the visualizer canvas and can be retrieved by clicking.
The ? button is used to check additional information for the use of the chosen function.
The Keyboard Shortcut key is available in Structure Builder. The Keyboard makes it simple to operate.
This is a list of file formats that can be uploaded/downloaded to structure builder.

Special quasirandom structures (SQSs) are an important tool in modeling disordered alloys with atomic resolution.
Define supercell size of x, y and z axis.
Define group range for random distribution. (Automatic value is the mean value of both 2nd and 3rd nearest neighbor distance)
Define atomic fraction in an individual site. (Caution! When the atomic fraction is converted to # of atoms, # of atoms should be an integer.)
This value is the maximum simulation time and should be defined due to the stable simulation. (In the case of 96 atoms, the maximum simulation time is recommended to be 24 hours. In the case of 48 atoms, the maximum simulation time is recommended to be 6 hours.)
Structure Builder consists of the visualizer menu that displays the structure and the manipulator menu for manipulating structures.
 Watch the Modeling Clip
Watch the Modeling Clip
The simplest way to create a simulation model is to upload a structure file.
Drag and drop a file to Visualizer or click on the
icon to upload a file. You can upload most file types, such as CIF, POSCAR, PDB, xyz, and so on. Alternatively, you can add structures using the 'Modelling' tab menu.

|
Crystal: Bring the primitive cell information from Materials Project.
Chemical: Bring the molecule structure from PubChem. |

|
It gets a structure data from the previously performed simulation jobs. Click on Init/Final to add the initial/final structure to the Structure list. |

|
It loads a structure data from the module that currently exists on the work page and adds it to the structure list. Furthermore, it can only load from the modules that contain structure data, such as Quantum Espresso, Structure Builder, and others. |
|
|
It loads a structure from the file and adds it to Visualizer. It supports almost of formats, such as CIF, POSCAR, PDB, and XYZ. |
|
Build common bulk crystal by setting "crystal system", "lattice constant", and "element" information |

|
We have pre-added structures that are used frequently. You can add graphene unit cells and Silicon bulk unit cells. |

|
It enables adding a molecule in 3D by drawing the structural formula. It performs optimization using the MMFF94 Forcefield of Open Babel. |

|
You can create a structure by entering coordinates directly, or you can modify a structure by modifying the coordinates. You can also copy and paste text containing structure information. |
You can use several functions by manipulating the mouse on the structure canvas. Moreover, you can use the mouse wheel to zoom in/out or drag to rotate the time point. Right-click to use the following functions:
| Background | Selects a background color for the Visualizer canvas |
| Perspective | Adjusts perspective |
| Atom | Adjusts the atom size |
| Bond | Adjusts the bond thickness |
| Shift | Shifts atoms to identify structures; does not change the actual coordinate information |
| Cell | Determines whether the cell line should be displayed |
| Info | Determines whether the cell information should be displayed. (right-bottom side)
Off: Do not display any information Crystal: Display information of periodic cell Molecular: Display molecular information |
| Axis | Determines whether the axis arrow should be displayed |
| Select Info | Determines whether the information should be displayed when an atom is selected |
| Ghost | Display unit cell repeatedly. Even there are only a few atoms, the program accepts them as an infinitely repeating structure. |
| Light | Sets the angle and intensity of the light emitted to structures |
| Advanced | You can adjust the quality and texture of the 3D model. Decreasing 'Representation' or 'Resolution' helps render for the low-memory environment. |
| Select | Turns the selection mode on or off; can operates viewpoint using the mouse if you press and hold the Space in the Select mode | ||||||||||||||||
| Mode | Specifies the shape of the area will select
|
||||||||||||||||
| Method | Determines options that will be added to the selection when another atom is selected after the selection
|













| Download | Downloads the structure currently present in the visualizer canvas in the selected file format |
| Distance | Selects two atoms to find out the distance |
| Angle (Vectors) | Selects three atoms to find out the angle |
| Angle (Planes) | Selects four atoms to find out the angle of the plane |
| Space Group | Calculates the space group of the structure; takes longer time for a large structure as it involves a large amount of computation |
MatSQ provides a variety of tools to manipulate the atomic structure. You can also use the Manipulator menu to add or move atoms, resize cells, or clone unit cells to create supercells. Up to 10 Manipulate details are stored in the history at the bottom of the visualizer canvas and can be retrieved by clicking.
The ? button is used to check additional information for the use of the chosen function.

|
Compresses or expands a structure by applying a strain based on the origin.
You can add the changing structures with entered range and interval on the 'Range' tab. The structures are added to the Structure list . |

|
Clones a structure using an integer multiplication factor |

|
Adds a free space that is recognized as a vacuum
To build a surface, you must give at least 10 Å of vacuum on one axis. |

|
Cleaves the crystal according to the Miller index (cleavage) |

|
Merges two structures out of the structure list |

|
It finds a primitive cell, the minimum repeating unit of the structure. |
|
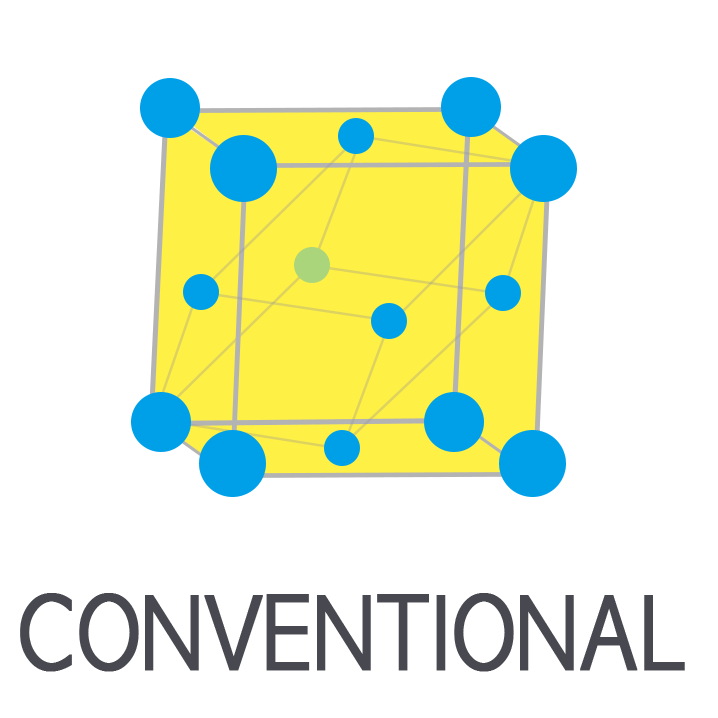
It generates the conventional cell from the primitive cell. |

|
Adds an atom to a structure |

|
(Add Molecule) You can add molecules or specific structures.
(Fill) You can fill atoms, molecules, or specific structures randomly inside to cell. |

|
You can fill structures randomly. Import structures from the Modeling module or structure list . The cell size will be expanded to prepare sufficient space. |

|
(Move) Moves the selected atom
(Flip) Flips the selected atom symmetrically to create a mirror-image structure (Inside) If there is an atom outside the cell, the Inside function brings the atom inside, taking account of the periodicity. |
|
Rotates the selected atom |

|
Replaces the selected atom with another element |

|
Creates a NEB (Nudged elastic band simulation) image; includes the initial and final images in the image count |

|
For high throughput calculations, the function creates combinatorially generated structures by replacing the selected atom with another element and add to Structure list. (The number of Elements The number of selected atoms ) Watch out for memory deficiency. |
|
Finding available conformers. The generated structures are added to the Structure list . |
The Keyboard Shortcut key is available in Structure Builder. The Keyboard makes it simple to operate.
| Function | Key | View mode | Select mode |
|---|---|---|---|
| Select | Space | (hold down) Enable mouse control |
|
| s | Open select mode | ||
| Esc | Exit select mode | ||
| r | Exit select mode | ||
| Delete | Delete (Mac: Fn+Backspace) | Delete the selected atoms | |
| Move & Rotate | ↑ | Move up the selected atoms in the viewport (1 Å) |
|
| ↓ | Move down the selected atoms in the viewport (1 Å) |
||
| ← | Move left the selected atoms in the viewport (1 Å) |
||
| → | Move right the selected atoms in the viewport (1 Å) |
||
| q | Rotate selected atoms around the y axis (left) |
||
| e | Rotate selected atoms around the y axis (right) |
||
| Viewport | x | View YZ plane | View YZ plane |
| y | View XZ plane | View XZ plane | |
| z | View XY plane | View XY plane | |
| a | View BC plane | View BC plane | |
| b | View AC plane | View AC plane | |
| c | View AB plane | View AB plane | |
| [ | Rotate left the viewport around the z-axis (10˚) |
Rotate left the viewport around the z-axis (10˚) |
|
| ] | Rotate right the viewport around the z axis (10˚) |
Rotate right the viewport around the z axis (10˚) |
|
| { | Rotate forward the viewport around the x axis (10˚) |
Rotate forward the viewport around the x axis (10˚) |
|
| } | Rotate backward the viewport around the x axis (10˚) |
Rotate backward the viewport around the x axis (10˚) |
|
| f | Viewpoint flip | Viewpoint flip | |
| Group | Shift + 0~9 | Grouping selected atoms to group #n (until refresh) | |
| Numeric keys (0~9) | Loading selection of the group #n | Loading selection of the group #n (until refresh) |
This is a list of file formats that can be uploaded/downloaded to structure builder.
Molecule Builder is modeling tool for creating single-molecule and polymers. To obtain 2D to 3D conversion and optimization, it use molecular mechanics force field (MMFF94) via Open Babel.


Draw the molecules or polymer unit in 2D structural formula form. For the detail, please refer to the following posts.
 button to start the 3D convert. The
button to start the 3D convert. The  icon will appear when after the convert is finishing. Please note that the convert job will be submitted to the cloud server if the number of atoms is numerous.
icon will appear when after the convert is finishing. Please note that the convert job will be submitted to the cloud server if the number of atoms is numerous.
You can check the progressing calculations at the cloud server on the dashboard▶ of the top navigation bar. The running job is marked as the icon. After finishing, update the job status by clicking the button.
icon. After finishing, update the job status by clicking the button.

The 3D model converting result can find on the 'Visualizer' tab. Adjust the appearance settings, or measure the interatomic distance and angle on the right-clicking setting window. You can download the structure in the format you desired.
|
|
It brings the optimized model from the chemical open database (PubChem). You can search the model by IUPAC name or common name.
The imported model is listed on the right side. Click on the list to see the 2D structure. You can also instantly view the 3D model from the "Visualizer" tab. If you want to newly optimize the showing model, click the Polymer copy icon which appears when hovering the mouse pointer on the list. |
|
|
You can upload the structure file. You should optimize manually when you uploading the mol file. If you upload extension files such as cif, xyz, etc.(except mol file), you can immediately see the 3D model in the "Visualizer" tab. If you want to newly optimize the showing model, click the Polymer copy icon which appears when hovering the mouse pointer on the list |
|
You can find the conformational isomers of the molecule, which was drawn as the 2D structure formula. The searched conformers are added to the right list, and the relative energy data will be written together. It will be added only the stable top 10 structures if the number of found conformers is numerous. For further information, please refer to the [MatSQ Tip] How to find stable molecule geometry: Conformational Isomer post. |
|
This menu enables the substitution of R Group to diverse chemical groups at one time. |
|
Find the duplicates of the generated molecules list. The duplicates are selected automatically. Click the button to delete all the selected duplicates. |
|
|
It searches for structures with high chemical similarity using the Tanimoto coefficient. It is the same function as the similar molecule search service provided by PubChem. |
Draw the molecules or polymer unit in 2D structural formula form. For the detail, please refer to the following posts.
- [MatSQ Tip] Molecule Modeling in Materials Square
- [MatSQ Tip] How to model complicated molecule
- [MatSQ Tip] How to Model Polymer in 3D: Molecule Builder
button to start the 3D convert. The You can check the progressing calculations at the cloud server on the dashboard▶ of the top navigation bar. The running job is marked as the
The 3D model converting result can find on the 'Visualizer' tab. Adjust the appearance settings, or measure the interatomic distance and angle on the right-clicking setting window. You can download the structure in the format you desired.
Special quasirandom structures (SQSs) are an important tool in modeling disordered alloys with atomic resolution.
Define supercell size of x, y and z axis.
Define group range for random distribution. (Automatic value is the mean value of both 2nd and 3rd nearest neighbor distance)
Define atomic fraction in an individual site. (Caution! When the atomic fraction is converted to # of atoms, # of atoms should be an integer.)
This value is the maximum simulation time and should be defined due to the stable simulation. (In the case of 96 atoms, the maximum simulation time is recommended to be 24 hours. In the case of 48 atoms, the maximum simulation time is recommended to be 6 hours.)